Article Text

Abstract
Background In the clinically and genetically heterogeneous group of the hereditary spastic paraplegias (HSPs), mutations in the SPAST gene are most frequently found and cause a pure autosomal dominant form.
Objective To provide the clinical and genetic characteristics of Dutch patients with HSP due to a SPAST mutation (SPG4).
Methods SPAST mutation carriers were identified through a comprehensive national database search. Available medical records were reviewed.
Results 151 mutation carriers carried 60 different changes in the SPAST gene, of which one was a known polymorphism, and 27 were novel. Missense mutations were most frequently found (39%). Clinical information was available from 72 mutation carriers. Age at onset ranged from 1 to 63 years with a bimodal peak distribution in the first decade and above age 30. The predominantly pure spastic paraplegia was accompanied by deep sensory disturbances and sphincter problems in almost 50%. An additional hand tremor was found in 10%. Patients with missense mutations and exon deletions did not reveal a distinctive phenotype.
Conclusions Dutch SPAST mutation carriers show a broad mutation spectrum, with 27 novel mutations in the present series. A bimodal peak distribution in age at onset was found and an accompanying tremor as peculiar feature of SPG4. The pathogenicity of S44L, the first exon 4 mutation, and a possible autosomal recessive mode of inheritance are discussed.
- SPAST
- novel mutations
- tremor
- exon 4
- S44L
- genetics
- heredit spastic paraplegia
- tremor
Statistics from Altmetric.com
Introduction
The hereditary spastic paraplegias (HSPs) constitute a genetically and clinically heterogeneous group of disorders of which the main clinical feature is progressive lower-limb spasticity due to pyramidal tract dysfunction. The cardinal signs result from a ‘dying back’ degeneration of the corticospinal tracts and dorsal column, predominantly affecting axonal transport of the longest fibres that innervate the lower extremities.1 Neuroimaging of HSP patients can reveal spinal cord atrophy, mostly at the thoracic level.2 In addition, corpus callosum atrophy (although more common in autosomal recessive HSP), atrophy of the cerebellum and white-matter lesions have all been found in HSP.3 4
If neurological signs are limited to the lower limbs, eventually accompanied by urinary urgency and mildly impaired vibration sense at the ankles, HSP is classified as ‘pure.’5 In contrast, HSP is classified as a ‘complicated’ form if additional neurological signs are present, such as mental retardation, extrapyramidal signs, visual dysfunction, epilepsy or systemic involvement.5
HSP may be inherited as an autosomal dominant (AD), an autosomal recessive (AR) or an X linked disease, with more than 40 loci identified.6 AD transmission is observed in 70 to 80% of all HSP cases in Western countries.7 Mutations in SPAST are responsible for about 40% of the AD-HSP cases and cause a predominantly pure HSP.4 Over 150 mutations of different types (missense, nonsense and splice mutations, deletions and insertions) along the SPAST gene have been reported. SPAST is a gene encoding the spastin protein, which is a member of the ATP-ases Associated with various cellular Activities (AAA) family.4 Both the AAA domain and the MIT(microtubule interacting and trafficking) domain of the SPAST gene play an important role. Recent studies confirmed that spastin possesses microtubule-severing activity, necessary for axonal transport.8 A loss of function of spastin due to a SPAST mutation could thus lead to axonal dysfunction.
There is a broad clinical spectrum of SPAST mutations, even within families, and the genotype–phenotype correlations remain largely unclear. To further expand our knowledge on the phenotypic and genetic spectrum of SPAST-linked HSP, we studied the mutations and disease characteristics of a comprehensive cohort of Dutch SPAST mutation carriers and found several new features.
Methods
Patients
The DNA diagnostic laboratory within the Department of Human Genetics of the Radboud University Nijmegen Medical Centre (RUNMC) is the single national laboratory to provide SPAST mutation analysis for The Netherlands, being offered since 2000. Thus, we were able to identify all Dutch SPAST mutation carriers from a laboratory information system query. Available medical records and imaging data were reviewed. A clinicogenetic HSP-database was produced, containing clinical information from history, with the age at onset as mentioned by the patient, and neurological examination, combined with the results of genetic testing and additional investigations (CT-MRI-EMG-laboratory-other). The study has been carried out in The Netherlands in accordance with the applicable rules concerning the review of studies by research ethics committees and informed consent.
Genetic analysis
Mutation analysis of the SPAST gene was performed by sequencing of the coding sequences including flanking intronic sequences as well as multiplex ligation-dependent probe amplification (MLPA) assay in all patients, using the methods described previously.9 10 NM_014946.3 was used as reference sequence, with nucleotide 1 corresponding to the A of the start codon. If the results were indicated by multiple flanking probes within the MLPA test, they were considered as indicative for a true deletion. Test results were confirmed by an independent test according to standard procedures. Determination of pathogenicity of point mutations was obtained by an in silico approach using the prediction programs SIFT (Sorting Intolerance from Tolerance; http://sift.jcvi.org), Align GVGD (http://agvgd.iarc.fr/) and POLYPHEN (Polymorphism Phenotyping; http://genetics.bwh.harvard.edu/pph/).11 12
Results
From July 2000 to July 2008, a mutation analysis was performed in 1386 Dutch patients with suspected HSP, in the presence or absence of affected family members. This yielded 151 (ie, 11%) positive carriers originating from 84 families. Five patients were sporadic. We had sufficient clinical data from 72 carriers (from 47 families) to study the phenotypic spectrum.
Genotypic spectrum
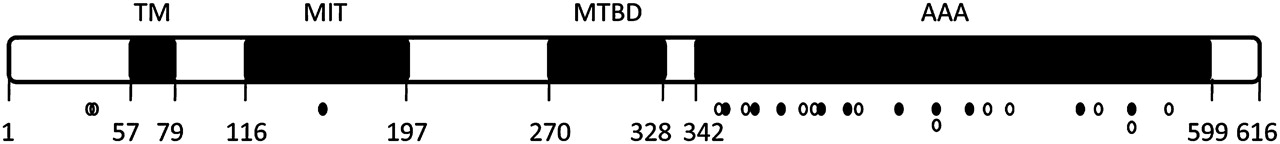
The 151 mutation carriers originating from 84 families were found to carry 60 different changes in the SPAST gene, one of which is a known polymorphism (p.Ser44Leu; see below and Discussion). Overall, we found 23 missense mutations (39%), 10 splice site mutations (17%), nine small deletions (15%), eight deletions of single or multiple exons (14%), six nonsense mutations (10%), two duplications (3%) and one insertion (2%) (supplementary table 1). Mutations occurred throughout the whole gene. According to the international electronic database (http://www.hgvs.org), we identified 27 novel SPAST mutations (table 1). By MLPA analysis, we identified eight different large exonic deletions in the SPAST gene, three of which are novel. Most families showed private mutations. Some mutations were observed more frequently, like c.1174del (p.Ala392fs). All but one novel missense mutations are clustered in the AAA domain. The other mutation is located in the MIT (microtubule interacting and trafficking) domain (figure 1).
List of 27 novel mutations identified in the SPAST gene in the hereditary spastic paraplegia cohort

Schematic figure showing the structural domains of SPAST, showing all missense mutations identified in this study. Open circles indicate known mutations, and closed circles indicate novel mutations. All but one novel missense mutation were detected in the ATP-ases Associated with various cellular Activities (AAA) domain; the other mutation was detected in the Microtubule Interacting and Trafficking (MIT) domain. MTBD, MT, microtubule binding domain; TM, transmembrane domain.
Phenotypic spectrum
The phenotype could be defined for 72 cases. Five of these were asymptomatic SPAST mutation carriers (according to their history) and were not included in determining the phenotypic spectrum, apart from the tendon reflexes (table 2). In total, 47 distinct families could be identified based on available family information.
Phenotypic characteristics in 72 SPAST mutation carriers, of which five were asymptomatic according to history
The family history of these patients revealed an autosomal dominant mode of inheritance with certainty in 72% of the 72 cases. Five cases were sporadic (6.9%). In one family, an autosomal recessive mode of inheritance was observed, associated with a unique missense mutation in exon 14 (c.1600C→G: p.Leu534Val). The two affected siblings, who showed a pure spastic paraparesis, were homozygous for the mutation, while the heterozygous consanguineous parents were fully asymptomatic at ages of 73 and 68 years (see also Discussion).
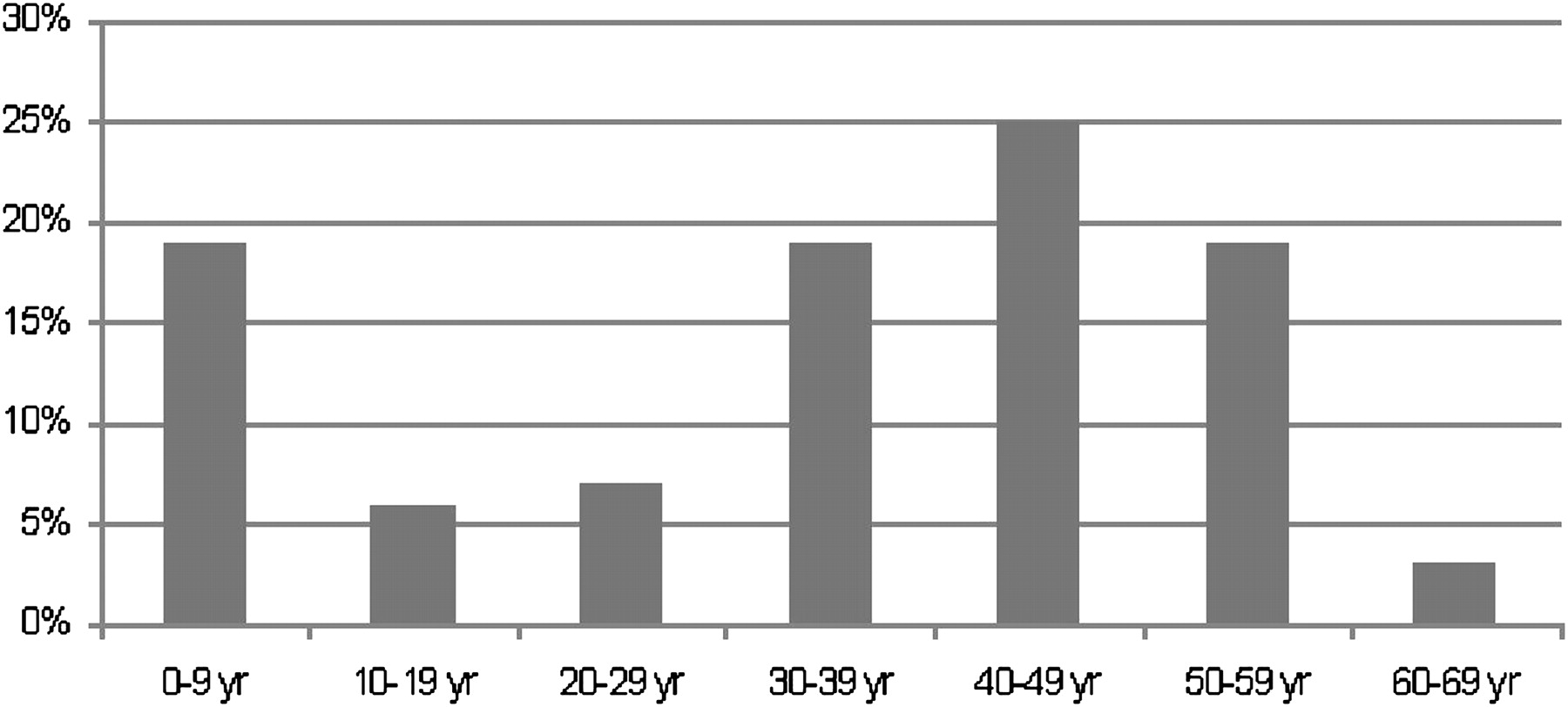
The age at onset varied from 1 to 63 years (mean 33.4±SD 18.3 years), and showed a bimodal distribution with a first peak in the first decade and a second peak between 30 and 60 years (figure 2).

{kind=link}
{kind=link}
Distribution (%) of the age at onset in 67 hereditary spastic paraplegia patients with SPAST mutations, with a peak of 19% with an age at onset before 10 years and a peak of 25% with an age at onset between 40 and 50 years.
Most patients presented with gait difficulty (table 2). The overall phenotype was that of a slowly progressive, mostly pure spastic paraparesis with only little or no arm involvement. The strength and tone of the upper limbs were mostly normal, but the tendon reflexes in the arms were brisk in 61%. Mild cerebellar ataxia of the arms was present in a few cases. Ataxia of gait was more common; 32% of the patients presented with a spastic ataxic gait and/or difficulty with tandem gait. Muscle tone of the lower limbs was increased in most patients (85%). Muscle strength was only mildly impaired or normal, with 80% of cases who demonstrated strength of at least MRC 4. The small group with a (lower limb) muscle strength of less than MRC 4 showed a significant longer disease duration (mean of 24 years) compared with the group with a strength of at least MRC 4 (mean duration of 10 years). Brisk reflexes of the lower limbs were found in 95% with Babinski signs in 86% (in 72% bilaterally). About one-third of the patients used some sort of walking aid, and 15% were completely wheelchair bound.
Deep sensory modalities were disturbed in almost half of the patients (47%). Vibration sense was predominantly impaired. Bladder disturbances, predominantly urinary urgency, were present in 42% of the patients, and anal sphincter disturbances in 15%.
Swallowing problems were mentioned by two patients (3%), in whom mild dysarthria was also noticed.
Patients with missense mutations did not show a significant earlier age at onset (33.8 years vs 33.3), compared with other types of mutations. In both the <35 age group and the >35 age group, we observed an approximately 40% proportion of missense mutations, 38% and 43% respectively, without a preference for the younger age at onset group. The clinical presentation and the age at onset of the patients with an exonic deletion were similar to those with loss-of-function mutations, that is, missense, nonsense and splice site mutations.
Neuroimaging of the brain and the spinal cord was performed in 39 patients. Eight (21%) of these scans showed HSP-associated abnormalities: four patients had atrophy of the spinal cord; the others had atrophy of the cerebrum (n=1), cerebellum (n=1) and a thin corpus callosum (n=2). Unspecific white-matter lesions (WML) were seen in four (10%) patients, who were at that time aged 44–62 years.
Complex phenotype
Over one-fifth (22%) of the patients presented with a relatively mild complex phenotype (table 3), based on clinical and/or imaging features. The abnormalities were mostly minor. A mild tremor of the arms was present in 7/67 patients (10%). Three patients showed a postural tremor, three patients an intention tremor and one patient an action tremor. A more severe complex phenotype, based for example on the presence of mental retardation or dementia, was rare.
Pure versus complex phenotype of the 67 SPAST patients based on clinical and/or imaging features
Clinical data of the S44L family
We found one family in which the known p.Ser44Leu polymorphism is segregating. No additional pathogenic SPAST mutation was identified. The mother was heterozygous for this variant and showed no signs or symptoms. The father was also heterozygous but presented with sphincter problems at the age of 5 years. On examination, his legs were hypertonic with increased tendon reflexes and Babinski's sign bilaterally. The son, who was homozygous for the p.Ser44Leu variant, presented with sphincter disturbances at age 6, but without any difficulty of gait or abnormalities on examination (see also Discussion).
Discussion
SPAST mutations comprise by far the most frequent form of autosomal dominant spastic paraplegia (between 15 and 40% of AD-HSPs).4 13 Our cases represented 11% of all requests for SPAST mutation analysis, but it should be kept in mind that these requests emerged from an unselected cohort that included sporadic and even probably recessive paraplegia cases. A German study revealed 17% SPAST mutations in a comparable population suffering from spastic paraplegia with or without a family history.9
Here, we report a comprehensive overview of the phenotypic and genotypic spectrum of all identified SPAST mutation carriers in The Netherlands. Since the RUNMC is the only centre in The Netherlands providing SPAST mutation analysis, this overview includes all known SPAST mutation carriers in The Netherlands. The identification of 151 SPAST mutation carriers results in a prevalence of 0.92 carriers per 100.000 in The Netherlands.
Mutations
Of the 60 different changes in the SPAST gene identified, one was a previously described non-pathogenic polymorphism (p.Ser44Leu), and 59 were true mutations. We also confirmed the existence of exonic deletions, which is consistent with findings recently reported.13 14 In our cohort, missense mutations were the most frequent (39%), followed by splice site mutations (17%) and small deletions (15%). Comparable figures were found in two other studies.15 16
The most frequent mutation was a previously described mutation (c.1174del), in the AAA domain.16 This deletion results in a frameshift after the alanine at position 392 and was found in five out of 84 families (6%).
A novel finding is the deletion of exon 4 in one family suffering from a well-defined spastic paraplegia. The pathogenicity of this mutation is not certain, as SPAST is alternatively spliced. Exon 4 is spliced out in one transcript, coding for a slightly shortened isoform (NM_199436.1). The transcript without exon 4 is still in frame, and it is not known what the functional consequences of this deletion are.
Mode of inheritance
In 7/84 (8%) families, the mode of inheritance was uncertain but probably best fits with incomplete penetrance of an autosomal dominant trait, or incomplete clinical assessment of the parents. Incomplete penetrance and reduced or delayed expression, which is depending on the age at the time of examination, have been described before in a relatively high proportion (24.1%) of SPAST mutation carriers.17 In this Irish study, even a case of true non-penetrance was observed.
Five cases seemed sporadic (6.9%), due to a de novo mutation, incomplete penetrance, somatic mosaicism, non-paternity or incomplete clinical assessment of the parents. De novo mutations have been found in 6% of cases in an Italian study and in 12% in a French study.18 19 As we have not systematically investigated all family members, we cannot comment further on our sporadic cases.
One family presented with a (pseudo-)recessive inheritance. Both parents, who were consanguineous, had a heterozygous missense mutation, c.1600C→G (p.Leu534Val), without signs or symptoms at ages 73 and 68 years, respectively. Their two children, homozygous carriers of the mutation, presented with a pure spastic paraparesis, both with an onset at age 39 years. Both patients were tested negative for SPG7. There are several explanations for these findings. First, this mutation (c.1600C→G, p.Leu534Val) may only be pathogenic in a homozygous state (thus representing a recessive disease). Second, the opposite SPAST allele may in fact be (partly) deleted. The allele carrying the c.1600C→G mutation would thus appear to be homozygous after sequencing. This, however, is very unlikely, as both parents are carrier of the mutation. The third explanation is that the mutation is a non-pathogenic polymorphism. In that case, the diagnosis cannot be confirmed genetically. However, a different heterozygous missense mutation affecting the same amino acid residue, c.1601T→C (previous nomenclature in literature: c.1726T→C), p.Leu534Pro, has been described causing an autosomal dominant pure HSP.20 The leucine-to-valine change (both non-polar, hydrophobic amino acids) is less dramatic than the leucine-to-proline change (a moderately polar, negatively charged amino acid with the additional capacity to form a hydrogen bond). This could well explain the fact that L534P acts in a dominant way, whereas L534V only has a pathogenic effect when present in a homozygous state. The p.Leu534Val concerns a conservative change of a strongly conserved amino acid. Due to the high conservation, the change is predicted to affect protein function by online prediction tools SIFT and Align GVGD.1 2 Also, the fact that p.Leu534Val is located in the AAA domain may cause it to be pathogenic.11 12
S44L
S44L (p.Ser44Leu) is a relatively rare but well-described non-pathogenic polymorphism. In a North American control population, the L44 allele was found in 0.6% of individuals examined and in a British control population even in 3.1%.15 21 However, a role as a phenotypic modifier is also attributed to S44L previously.15 21
One family in which this polymorphism is segregating was found in our study. The father's disease, which is compatible with an early-onset spastic paraplegia, may be caused by an as yet unidentified mutation in one of the genes underlying paraplegia, suggesting a role for p.Ser44Leu as a genetic modifier. This may include an unidentified mutation in a regulatory or intronic region of SPAST. An English severely affected child with compound heterozygosity for p.Pro361Leu and p.Ser44Leu in the SPAST gene supports this hypothesis.22 Urinary urgency and frequency together with hyper-reflexia were the main features in this family, comparable with our case. In case of co-occurrence of another SPAST mutation, S44L would act via a gain of function mechanism and worsen a primary ‘loss of function’ effect caused by a different loss of function mutation occurring in trans.23 An imbalance between short and long isoforms could be responsible for a mild pathogen effect of an isolated S44L mutation.23 Another explanation could be that p.Ser44Leu shows a high variability in phenotypic expression, rather than being a genetic modifier. Two Norwegian families with HSP and a p.Ser44Leu polymorphism were described, both without an identified pathological SPAST mutation.24 Some of the family members were homozygous, others heterozygous for the polymorphism, associated with or without clinical symptoms. Development of a more severe disease due to a homozygous mutation of S44L, as in the son in our case, has been suggested before.16
Thus, rather than being completely non-pathogenic and innocent, it should be suspected that p.Ser44Leu, under specific conditions, may cause a mild HSP phenotype, with a more severe phenotype, when combined with a classical SPAST mutation, or in a homozygous mutation.
Phenotype
The phenotype of gait difficulty due to a slowly progressive pure spastic paraparesis with little or no arm involvement, accompanied by impairment of deep sensory modalities and sphincter problems, is fully consistent with the phenotype as reported in the literature.15 16 Compared with almost 42% bladder disturbances and 14.5% anal sphincter disturbances in our cohort, urinary urgency was found in only 21% of the British cases.15 This difference may be caused by the intensity of questioning rather than by a true difference. Also, the proportion of patients who were wheelchair-bound in our study (15%) is comparable with the 17% reported previously.4 In contrast, swallowing problems and dysarthria, which were present in two of our 67 patients with symptoms (3%), have not been reported previously.
One-fifth of our patients had a mostly mild, complex phenotype, with additional signs or symptoms not attributable to the pyramidal tract or dorsal columns. Hand tremor was observed in 10% of SPAST patients. A literature search did not reveal any recent reports of tremor in SPAST patients but might have been overlooked clinically. In a family description from 1963, a ‘slight intention tremor’ in four patients was mentioned as an atypical feature of AD-HSP.25 The other additional neurological symptoms or signs found in a part of our cohort, such as neuropathy, epilepsy, mental retardation, cerebellar atrophy and thinning of the corpus callosum, have previously been described in SPAST patients.6 A mild form of dementia, which has been described as an associated symptom in SPAST patients, was however found in only one of our SPAST patients.26 Based on neuropsychological testing, CSF findings and brain MRI in our patient, this cortical dementia could be either SPAST-related or Alzheimer's dementia.
The mean age at onset in our cohort was 33.4±18.3 years, ranging from 1 to 63 years, with a bimodal distribution showing a peak in the first decade and a second peak in the fourth to sixth decade. A mean age at onset of 29–34 years with a comparable broad range was found in previous studies.15–17 However, a clear bimodal distribution, as we describe, was not found in these European studies, and we clearly found more patients with an age at onset in the first and sixth decade (both 19%), compared with other reports. The challenge here is to assess the exact age at onset. Currently, this is estimated by careful history taking, but this method may yield systematic error, as many patients probably go through an initial phase in which they do not yet experience symptoms, while hyper-reflexia could already be present.
In the group with exonic deletions, a similar phenotype was found in our study, as in the group with loss-of-function mutations caused by a base-pair substitution or a small deletion or insertion, as has been described previously.13 14 In contrast to other studies, our results do not confirm the recently proposed hypothesis of an earlier onset of the disease when caused by a missense mutation in the AAA domain.10
Spinal cord atrophy, as was found in four of our patients, has been described in SPAST patients.2 In the same study, MRI of the brain was normal, and the thickness of the corpus callosum did not differ from healthy controls.2 Another study also showed atrophy of the midthoracic cord, and in this study the corpus callosum was indeed significantly smaller in patients than in healthy controls.3 However, in that study, not all patients were tested for SPAST mutations but were merely diagnosed as having pure HSP. On the contrary, another study states that a thin corpus callosum is not associated with SPAST-linked HSP.27 We found two patients with a thin corpus callosum, supporting a possible association with SPAST.
Four patients showed so-called unspecific WML on brain-MRI. It is questionable whether this finding is of any value, that is, linked to HSP. It may be related to age, as the youngest patient with WML was 44 years old. The literature is also ambiguous. In some, mostly older papers, WML are indeed linked to HSP.28 29 Others suggest that WML are not more common in HSP patients than in controls.30 No age-matched comparisons have yet been conducted.
Conclusion
Dutch patients with HSP due to a SPAST mutation show a broad mutation spectrum, with 59 different mutations identified, of which 27 are novel. Clinically, a predominantly pure spastic paraparesis was observed, with a wide range of age at onset, consistent with other reports of large populations of SPAST patients in Western countries.
Interestingly, a distinct bimodal peak distribution of age at onset, at the first decade and between 30 and 60 years of age, was found in the Dutch population. Compared with previous studies, an age at onset before 10 and after 50 years was relatively more frequent. Although urinary urgency is frequently described in the literature, bladder and anal sphincter disturbances were more common observed in this study and might have been underestimated. As a complicating feature, a tremor was found in almost 10% of the patients, a feature which needs more detailed investigation in future studies. The same applies to a possible autosomal recessive mode of inheritance of the missense mutation in exon 14 (c.1600C→G: p.Leu534Val), the role of the p.Ser44 Leu variant and of exon 4 mutations.
References
Footnotes
Scheffer H & van de Warrenburg BPC contributed equally to this study.
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.