Article Text

Abstract
Background Recurrent attacks in neuromyelitis optica spectrum disorders (NMOSDs) or myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD) can lead to severe disability. We aimed to analyse the real-world use of immunotherapies in patients with NMOSD and MOGAD, focusing on changes in treatment strategies, effects on attack rates (ARR) and risk factors for attacks.
Methods This longitudinal registry-based cohort study included 493 patients (320 with aquaporin-4 immunoglobulin G (AQP4-IgG) seropositive NMOSD (65%), 44 with AQP4-IgG seronegative NMOSD (9%) and 129 MOGAD (26%)) with 1247 treatments from 19 German and one Austrian centre from the registry of the neuromyelitis optica study group (NEMOS). We analysed unadjusted ARR and implemented survival analyses and Cox proportional hazard regression to assess efficiency and risk factors for subsequent attacks over time.
Results Rituximab and azathioprine are the most widely used immunotherapies in NMOSD as well as in MOGAD, with changes in distribution over the last decade. Immunotherapy demonstrated significant therapeutic effects in NMOSD but less pronounced effects in MOGAD. Risk factors for attacks included younger age and prior attacks under the same therapy. Efficacy varied among the different immunotherapies, with azathioprine, rituximab and eculizumab showing significant risk reductions in AQP4-IgG seropositive NMOSD.
Conclusions This study provides insights into the evolving treatment landscape and effectiveness of immunotherapies in NMOSD and MOGAD. Established off-label therapies continue to play an important role, especially for patients with stable disease, with emerging evidence supporting newly approved therapies. Future studies are needed to refine treatment algorithms and address the ongoing uncertainties in MOGAD management.
- NEUROIMMUNOLOGY
Data availability statement
Data are available upon reasonable request. The data that support the findings of this study are available from the corresponding author upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Effective attack prevention in neuromyelitis optica spectrum disorder (NMOSD) or myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD) is essential to avoid the accumulation of severe disability. Despite the existence of established off-label therapies, as well as data regarding the use of recently approved immunotherapies in clinical practice, there is a lack of information regarding their real-world application and comparison of effectiveness.
WHAT THIS STUDY ADDS
Traditional off-label therapies continue to be used, while the newer approved therapies are becoming increasingly important. Immunotherapies show significant effectiveness in aquaporin 4 immunoglobulin G (AQP4-IgG) seropositive as well as seronegative NMOSD. Effectiveness in MOGAD is less pronounced.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
This study highlights the importance and complexity of tailored immunotherapy approaches in managing rare diseases like NMOSD and MOGAD and underscores the need for personalised treatment algorithms and further studies.
Introduction
Neuromyelitis optica spectrum disorders (NMOSDs) and myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD) are rare neuroimmunological diseases.1 The phenotypes may overlap when the diseases manifest with the most common clinical symptoms of optic neuritis and/or longitudinal transverse myelitis. The detection of aquaporin-4 immunoglobulin G antibodies (AQP4-IgG) separates AQP4-IgG seropositive NMOSD from AQP4-IgG seronegative NMOSD.2–4 MOGAD has been characterised as a disease entity of its own by the presence of myelin oligodendrocyte glycoprotein immunoglobulin G antibodies (MOG-IgG).5 6
Recurrent attacks with incomplete recovery can lead to severe permanent disability and reduced quality of life in NMOSD and MOGAD.7–9 Based on case series and expert opinion, effective off-label therapies have been established to treat NMOSD, with rituximab becoming the treatment of choice in many countries.10–12 Recently, eculizumab, inebilizumab, ravulizumab and satralizumab have been approved for the treatment of AQP4-IgG seropositive NMOSD,13–17 but head-to-head comparisons among each other and with established off-label therapies are missing. The therapeutic strategies for MOGAD are even more uncertain due to limited evidence-based data and the absence of approved treatments. Evidence is growing that rituximab may be less effective in MOGAD,18 favouring alternative therapies such as intravenous immunoglobulins (IVIG) or interleukin-6 inhibition.19 20
Data on the effectiveness of immunotherapies in real-world settings may help to overcome these knowledge gaps, but are scarce and, unfortunately, mainly derived from mixed cohorts in which AQP4-IgG seropositive and seronegative patients with NMOSD and MOGAD were investigated together. Therefore, the present study was designed to analyse the change in treatments during the last decade, the effect of immunotherapies on attacks and attack-free survival and predictors of attacks from a large cohort of well-defined NMOSD and MOGAD subgroups from the registry of the Neuromyelitis Optica Study Group (NEMOS).
Methods
Study design and data collection
This longitudinal registry-based cohort study comprised data from 19 German and one Austrian NEMOS centres (www.nemos-net.de) between 1975 and database closure in October 2022; in cases with the last registry entry more than 365 days before the database closure, this entry was censored as the end of follow-up. Written informed consent was obtained from all patients before enrolment in the registry. Data were collected during regular clinical visits and included core demographic parameters (sex and year of birth), disease-specific characteristics (AQP4 and MOG-IgG serostatus, diagnostic criteria, year of manifestation and diagnosis), attacks (onset and clinical syndrome) and long-term treatment data (agent, start and end date). To ensure high-quality data, registry entries were exported and validated manually by two authors (AD and VH). Patients with a diagnosis of NMOSD according to the 2015 International Panel for NMO Diagnosis (IPND) criteria2 or a diagnosis of MOGAD based on the presence of MOG-IgG in serum and at least one attack with a typical clinical syndrome were included in the study (diagnosis and database entry preceded the diagnostic criteria proposed by Banwell et al 5). Standard testing for AQP4-IgG and MOG-IgG was performed with a cell-based assay (CBA)21 22 in most patients (for AQP4-IgG: 74% CBA, 22% unknown and 4% other than CBA; and for MOG-IgG: 80% CBA, 16% unknown and 4% other). Patients with insufficient core datasets or incomplete treatment data were excluded from the analysis.
Statistical analysis
Descriptive data were summarised by either median with range or frequency according to the type of the data and tested for differences by Kruskal-Wallis or χ2 test. We computed the unadjusted annualised attack rate (AAR) along with the corresponding 95% CI, assuming the number of attacks per year follows a negative binomial distribution. For further analysis, treatment episodes were defined as clinically meaningful stable treatment epochs, defined as the time between the first dose and either the assumed end of efficacy after the last dose (see online supplemental file for detailed information) or the last visit. For recurrent event analyses, these episodes were eventually split into separate periods at the date of attacks. Treatments with less than 30 cumulative patient years were excluded from the analysis. Survival analysis and calculation of attack-free rates were conducted using the Kaplan-Meier method, with concomitant steroid therapy considered a covariate in the subsequent analysis. To assess risk factors for attacks, we implemented a multivariate Cox proportional hazard regression model that included the patient ID as a cluster variable to account for the intraindividual correlation of observation. HRs and their 95% CI were computed. The p values <0.05 were considered statistically significant. Unless otherwise stated, all available episodes were included in the analysis of immunotherapies. All analyses were performed in R (V.4.3.1).
Supplemental material
Results
Description of the cohort
We screened 778 patients in the NEMOS database for eligibility (see online supplemental figure 1). After the exclusion of patients not fulfilling the 2015 IPND diagnostic criteria for NMOSD or not being classified as MOGAD or having incomplete or uncertain treatment data, 493 patients with 1247 treatments were included in the study. Of those, 320 patients were diagnosed with AQP4-IgG seropositive NMOSD, 44 with seronegative (AQP4-IgG/MOG-IgG) NMOSD and 129 with MOGAD (68.2% with >1 attack). Demographics varied significantly among the diagnostic groups but were typical for patients with AQP4-IgG seropositive and seronegative NMOSD and MOGAD (table 1). The median age at manifestation and diagnosis was higher in AQP4-IgG-positive NMOSD (43 and 48 years) than in double-negative NMOSD (35.5 and 39 years) and MOGAD (39 and 44 years). Similarly, the female sex constituted 88% of AQP4-IgG-positive patients with NMOSD, compared with only 40.9% in double-negative patients with NMOSD and 57.4% in patients with MOGAD. The most common syndrome at manifestation was transverse myelitis for AQP4-IgG-positive NMOSD (45.9%), followed by optic neuritis (35%). The occurrence of optic neuritis and myelitis in double-negative NMOSD was evenly distributed at 38.6% each, whereas in MOGAD, optic neuritis (45.7%) followed by myelitis (27.1%) was the most common manifestation. Other manifestations at disease onset were rare among all three groups. Comorbid autoimmune diseases were observed most frequently in AQP4-IgG-positive patients with NMOSD (35.3%), only rarely in double-negative patients with NMOSD (4.5%) and in 22.5% of the patients with MOGAD. Median follow-up time was 8.6 years in AQP4-IgG-positive NMOSD, 10 years in double-negative NMOSD and 5.4 years in MOGAD.
Patient characteristics
Immunotherapy characteristics and rates
Rituximab (348 treatment episodes in 301 patients for a total of 1110 patient years) and azathioprine (187 treatment episodes in 159 patients for a total of 678 patient years) were the most used immunotherapies in all three disease entities (online supplemental table 1). Similarly, rituximab, followed by azathioprine, was the most used first-line and second-line therapy. The choice of treatment was similar in AQP4-IgG seropositive and seronegative patients with NMOSD (rituximab 29.4% and 30.3%, azathioprine 13.7 and 13.8% of all treatment episodes, respectively). Looking at immunotherapies from the patient’s perspective, approximately one-third of patients across all three disease entities received azathioprine during the course of disease, which was applied first-line in the majority of cases (table 2). Rituximab, on the other hand, was applied in 68% of the AQP4-IgG seropositive patients with NMOSD (55% first line), whereas only 57% of the AQP4-IgG seronegative NMOSD (44% first line) and 45% of the patients with MOGAD (55% first line) received this immunotherapy. In accordance with the approval, new therapies were rarely used in AQP4-IgG seronegative NMOSD or MOGAD, but in AQP4-IgG seropositive NMOSD (6% eculizumab; 2% each eculizumab combination therapy, satralizumab and satralizumab therapy; 1% inebelizumab). Combination therapies other than those listed separately were more frequently used in patients with comorbid autoimmune diseases (18.8%) than in those without (6.0%). A detailed description of frequencies, sequences and treatment durations per treatment is provided in table 2 and online supplemental table 1.
Immunotherapies
Changes in the therapeutic landscape over time
The use of different immunotherapies between 2010 and 2022 is shown in figure 1, online supplemental figure 2 and table 2. The therapeutic landscape changed remarkably over time. In 2010, classical therapies used in multiple sclerosis (MS) accounted for as many as one-fifth (20.1%) of the treatments and rituximab for less than one-third (29.2%). The utilisation of rituximab, both first line and overall, increased significantly over time, peaking in 2018 (60.1% overall). The use of disease-specific newly approved therapies has steadily increased since the first approval in 2019, reaching 12.3% in 2022. Classical MS drugs were no longer prescribed in 2022.

Change of the treatment landscape in the Neuromyelitis Optica Study Group cohort. Stacked bar plot illustrating the distribution of treatments for each year. AQP4-IgG, aquaporin-4 immunoglobulin G; NMOSD, neuromyelitis optica spectrum disorders; MOGAD, myelin oligodendrocyte glycoprotein antibody-associated disease.
Looking at the different disease entities separately, the new therapies were only used in AQP4-IgG seropositive NMOSD as per approval, with a few exceptions. Interestingly, the use of rituximab decreased only slightly (61.3% in 2018, 55.0% in 2022) in AQP4-IgG seropositive NMOSD. Although first-line usage of rituximab is decreasing, it was still applied in 32% in the first line in 2022 (see online supplemental figure 2). However, in seronegative NMOSD, the proportion of rituximab decreased from its maximum of 73.1% in 2019 to 53.9%, in favour of azathioprine (30.8%) and tocilizumab (15.4%) in 2022. We observed similar effects for MOGAD, where the proportion of rituximab fell from 49.9% in 2018 to 37.3% in 2022.
Unadjusted efficacy of immunotherapies
The unadjusted AAR for all immunotherapies is presented in online supplemental table 1. The AAR for untreated episodes was 0.80 (95% CI 0.69 to 0.96); for AQP4-IgG seropositive NMOSD, it was 0.42 (95% CI 0.30 to 0.59) for seronegative NMOSD, it was 0.42 and 0.63 (95% CI 0.47 to 0.86) for MOGAD. We observed a trend towards higher AAR in AQP4-IgG seropositive NMOSD (1.32, 95% CI 0.90 to 1.96) as well as in seronegative NMOSD (1.36, 95% CI 0.82 to 2.28), not being evident in MOGAD (ARR 0.62 (95% CI 0.36 to 1.07)), for typical MS drugs in comparison to episodes with no treatment. Excluding classical MS therapies, the combined AAR for all drugs, compared with no treatment, was lower in AQP4-IgG seropositive NMOSD (AAR 0.48, 95% CI 0.41 to 0.56) as well as seronegative NMOSD (AAR 0.33, 95% CI 0.21 to 0.53) and remained nearly unchanged for MOGAD (AAR 0.67, 95% CI 0.52 to 0.88).
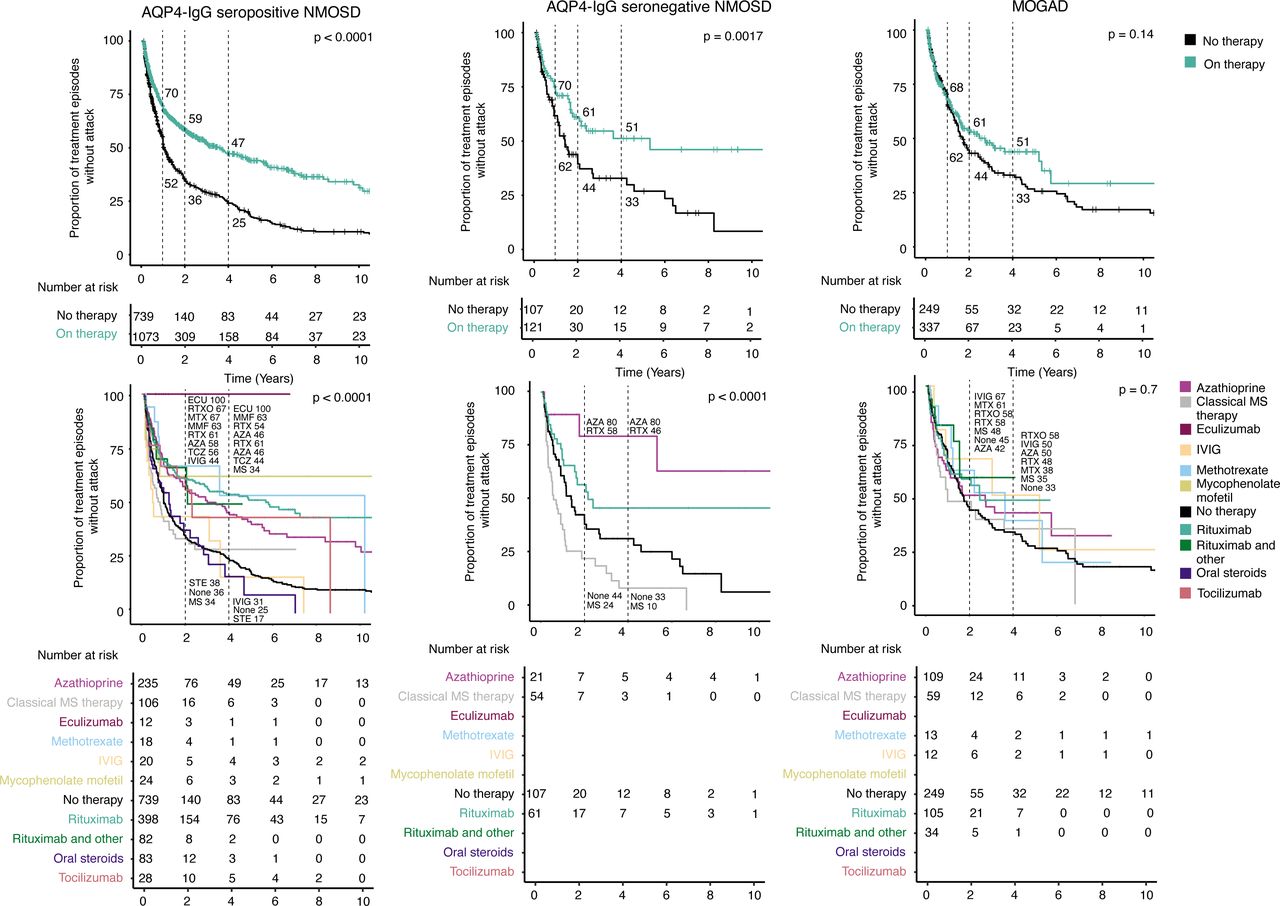
Regarding the risk of suffering subsequent attacks, recurrent treatment episodes were computed, splitting the 1247 available treatment episodes in the event of an attack. This resulted in 1750 therapy episodes, of which 219 were classical MS treatments. Together with 1095 episodes without maintenance treatment, these were subjected to a survival analysis using the Kaplan-Meier method. We compared episodes without maintenance therapy to all episodes with therapy except classical MS drugs (figure 2) and observed a robust therapeutic effect in both AQP4-IgG seropositive NMOSD and seronegative NMOSD, with an increase of attack-free episodes of +18, +23 and +22 percentage points (AQP4-IgG seropositive) and +18, +17 and +18 percentage points (AQP4-IgG seronegative) at 1, 2 and 4 years, respectively. For MOGAD, in turn, the effect was less pronounced, with only +2, +9, and +12 percentage points at 1, 2 and 4 years.

Unadjusted treatment effects in NMOSD and MOGAD. The unadjusted survival plots and their corresponding risk tables illustrate the risk for the next attack for all episodes (see methods for details). The dotted lines and the corresponding numbers highlight attack-free rates at 1, 2 and 4 years. The upper row compares any treatment (green, classic MS compounds excluded) with untreated episodes. The bottom row shows Kaplan-Meier curves for each compound separately. AQP4-IgG, aquaporin-4 immunoglobulin G; AZA, azathioprine; ECU, eculizumab; IVIG, intravenous immunoglobulins; MMF, mycophenolate mofetil; MOGAD, myelin oligodendrocyte glycoprotein antibody-associated disease; MS, multiple sclerosis; MTX, methotrexate; NMOSD, neuromyelitis optica spectrum disorders; RTX, rituximab; RTXO, rituximab and other; STE, steroids; TCZ, tocilizumab.
Substances with a sufficient cumulative treatment duration (>30 patient years) were then examined individually (figure 2). In AQP4-IgG seropositive NMOSD, we found that most treatments led to increased attack-free survival at 2 and 4 years, namely azathioprine +22 and +21, eculizumab +64 and +75, methotrexate +31 and +28, mycophenolate mofetil +27 and +38, rituximab without (+25 and +29) or with additional immunosuppressive therapy (+31 and +25) and tocilizumab +20 and +19 percentage points, at 2 and 4 years, respectively, whereas oral steroid therapy had no conclusive effect (+2 increase of attack-free survival at 2 years and decrease of −8 at 4 years). In seronegative NMOSD, azathioprine (+36 and +47 percentage points) and less pronounced rituximab (+14 and +13 points) improved the attack-free survival at 2 and 4 years. For MOGAD, however, the rates of attack-free survival of the different immunotherapies differed only slightly from untreated episodes at 2 years. Azathioprine (−3 and +17 percentage points) and rituximab (+13 and +15 points) had a weaker attack-preventive effect than in both NMOSD subgroups at 2 and 4 years. Unadjusted HRs for the different immunotherapies are provided in online supplemental table 3.
Risk factors for subsequent attacks and effectiveness of immunotherapies
A multivariate Cox proportional hazard model was used to assess the risk for attacks, adjusting for the cofactors sex, age at attack, line of therapy, concomitant steroid use and prior attack under the same treatment (table 3). Overall, the risk of an attack was lower in MOGAD than in AQP4-IgG seropositive NMOSD (HR 0.81, 95% CI 0.66 to 1.00, p=0.041), but the two NMOSD cohorts did not differ. Furthermore, we found that the attack risk decreased with age for all three subgroups (reference: <30 years; 30–50 years: HR 0.72, 95% CI 0.61 to 0.84, p<0.001 and >50 years: HR 0.50, 95% CI 0.41 to 0.61, p<0.001). A previous attack under the same immunotherapy increased the attack risk in AQP4-IgG seropositive NMOSD (HR 1.47, 95% CI 1.26 to 1.72, p<0.001) and MOGAD (HR 1.52, 95% CI 1.16 to 1.99, p=0.002), but not in AQP4-IgG seronegative NMOSD. Male sex decreased the attack risk in AQP4-IgG seropositive NMOSD (HR 0.74, 95% CI 0.59 to 0.93, p=0.010).
Risk for attacks in NMOSD and MOGAD
With regard to the effectiveness of immunotherapy, significant risk reduction for attacks was only seen in the largest group of AQP4-IgG seropositive NMOSD. The risk reduction was most pronounced for eculizumab monotherapy without any observed attacks (HR and CI not estimable, p<0.001). Furthermore, we observed a significantly reduced risk for rituximab (HR 0.66, 95% CI 0.51 to 0.86, p=0.002) and azathioprine (HR 0.72, 95% CI 0.51 to 1.00, p=0.049) in AQP4-IgG seropositive NMOSD. In seronegative NMOSD, risk reduction for azathioprine (HR 0.65, 95% CI 0.09 to 4.55, p=0.667) was not significant. For MOGAD, the HR was lowest for IVIG; nevertheless, it was not significantly reduced (HR 0.53, 95% CI 0.15 to 1.89, p=0.325), but only 13 patients were treated with IVIG. Interestingly, concomitant steroid therapy did not impact the risk reduction for all three entities (HR 1.04, 95% CI 0.72 to 1.49, p=0.848).
Treatment after failure of rituximab in AQP4-IgG-positive NMOSD
To identify treatments that were effective after rituximab has failed in AQP4-IgG-positive NMOSD, all immunotherapies used after a first attack while on rituximab for at least 60 days since its first dose were analysed using the Kaplan-Meier method (figure 3), irrespective of cumulative patient years. Attack-free rates after 2 and 4 years were higher for eculizumab (+36 and +46 percentage points) and for rituximab combined with another immunotherapy (+18 and +20 percentage points) compared with continuing rituximab monotherapy. Treatment with a monoclonal antibody against the interleukin-6-receptor (tocilizumab and satralizumab combined) did not show an effect (+3 and −2 percentage points attack-free survival). Using a multivariate Cox proportional hazard model, we observed a reduction in the risk of another attack for eculizumab only (p<0.001; online supplemental table 4).

{kind=link}
{kind=link}
{kind=link}
Unadjusted treatment effects after an attack under RTX in aquaporin-4 immunoglobulin G seropositive neuromyelitis optica spectrum disorders. The unadjusted survival plot and the corresponding risk tables illustrate the risk for the next attack under different treatments in episodes following an attack under RTX. The dotted lines and the corresponding numbers highlight attack-free rates at 1, 2 and 4 years. aIL6R, monoclonal antibodies against the interleukin-6 receptor (ie, tocilizumab and satralizumab); AZA, azathioprine; ECU, eculizumab; MMF, mycophenolate mofetil; MS, multiple sclerosis therapy; MTX, methotrexate; RTX, rituximab; RTXO, rituximab and other; STE, steroids.
Discussion
In our registry-based real-world cohort, we showed that immunotherapy has a significant therapeutic effect in AQP4-IgG seropositive NMOSD and seronegative NMOSD. This effect is less evident in MOGAD, however. We found that rituximab remains the most widely used immunotherapy in AQP4-IgG seropositive and seronegative NMOSD and MOGAD until 2022, demonstrating its continued valuation. Despite its off-label status, rituximab continues to be used not only in patients already receiving it for many years but also as first-line treatment in newly diagnosed AQP4-IgG seropositive patients. Possible reasons for this could be good long-term experience regarding efficacy and safety, as well as treatment costs.8
From the medical perspective, there is a lack of data from large cohorts that compare the effectiveness of currently available and recommended23 immunotherapies in AQP4-IgG seropositive NMOSD in a real-world setting. However, some studies showed that treatment with monoclonal antibodies with an assumed high efficacy was superior to classical immunosuppressant treatment in NMOSD.24–26 Although the numbers for immunotherapies other than rituximab and eculizumab were relatively small in our study, we could support this observation and validate these findings for NMOSD in our multivariate model for rituximab and eculizumab. However, recent data raised some safety concerns about fatal outcomes as well as attacks occurring in close temporal association with meningococcal vaccinations, which are mandatory for patients treated with complement inhibitors.27 Main concerns regarding anti-CD20 therapy include hypogammaglobulinaemia which may increase infection risks in vulnerable patients.28 Notably, only azathioprine, rituximab and eculizumab showed a significant risk reduction in AQP4-IgG-positive NMOSD in our cohort, which may be too small to prove the effectiveness of other drugs (particularly the other newly approved drugs, inebilizumab and satralizumab).
Regarding further attacks, younger age and a prior attack on the same therapy are risk factors in all three disease entities; female sex increased the risk only in AQP4-IgG seropositive NMOSD and MOGAD. The increased risk with continued therapy in the multivariate model supports the recommendation to switch to another drug as soon as possible, ideally to a different monoclonal antibody.23 29 In our cohort, eculizumab or add-on immunosuppressants to rituximab were therapeutic alternatives after rituximab failure in AQP4-IgG seropositive NMOSD. Surprisingly, add-on steroids failed to be protective, which has been suggested before30 and might be caused by the exclusion of short-term steroid therapies <60 days in our study. In addition, we may have missed the benefits of long-term concomitant steroid therapy in our patients because the doses administered may have been too low, which we cannot rule out because long-term steroid doses were not consistently recorded.
In addition to effectiveness in AQP4-IgG seropositive patients with NMOSD, we were able to validate the efficacy of immunotherapy in a truly, that is, AQP4-IgG and MOG-IgG seronegative, NMOSD cohort. Regarding the controversy as to whether AQP4-IgG seronegative NMOSD exists as a single entity or rather represents a heterogeneous subgroup of different diseases,31 it is crucial to recognise that many studies on this topic are probably confounded by the enrolment of patients with MOGAD.32 33 In our AQP4-IgG seronegative NMOSD cohort, we observed that azathioprine and rituximab were efficacious, although the effectiveness of rituximab was rather low in our multivariate model after correction for cofactors.30
The data on patients with MOGAD, on the other hand, is difficult to interpret. It is already known that the drugs used in NMOSD, particularly rituximab, are less effective.18 34 35 Our study, however, found an even higher AAR for rituximab compared with untreated patients. Only after correction for cofactors, the observed effect attenuated, persisting as less favourable compared with other studies. However, it should be noted that we did not compare the difference in AAR before and after the initiation of rituximab, as a number of studies indicated a considerable decrease in AAR after the initiation of rituximab. Furthermore, at least 20% of patients with MOGAD exhibit a monophasic course,5 36 depending on the observation period, which might also influence the effectiveness of immunotherapy. The risk factors for a relapsing or severe course are currently under discussion. Additionally, in MOGAD, it is common practice to not initiate immunotherapy after the first attack, likely introducing a bias towards more severe courses, but early oral steroid therapy in an adequate dose might be important.37 While it is beyond the scope of this manuscript to address this issue separately, further studies are urgently needed. Moreover, a thorough comparison with the currently preferred treatments IVIG and tocilizumab19 20 is still pending.
Limitations
This study has several limitations. The retrospective design may introduce biases related to data collection and selection, and, since most NEMOS centres are tertiary care centres, there might be a recruitment bias. Moreover, our cohort under-represents children, which is important in MOGAD. Regarding treatments, missing randomisation most likely leads to the assignment of treatments with a presumed higher effectiveness to patients with a more aggressive disease course and vice versa, which causes both overestimation and underestimation of the potency of the different treatments. Moreover, the lengthy observational period, spanning from 1975 to 2022, encompasses a period of significant change in diagnostic tools and criteria, as well as the treatment landscape. This complexity makes it challenging to interpret the results. While the study includes a large cohort from multiple centres, the sample size of some treatments is small. This is particularly true for the newly approved inebilizumab and satralizumab and therapies such as IVIG and tocilizumab to treat MOGAD. Finally, we did not assess safety or tolerability, both of which have an impact on treatment effectiveness.
Conclusion
This study provides valuable insights into the real-world effectiveness of immunotherapies in NMOSD and MOGAD in a changing treatment landscape. The newly approved therapies, as well as established off-label treatment, facilitate effective attack prevention in AQP4-IgG seropositive NMOSD. The future usage of these therapies with rituximab is still being most often applied, but with an obvious trend towards newer therapies, it indicates the need to further optimise treatment concepts. Further studies are needed to address the comparative effectiveness as well as the safety and tolerability of these immunotherapies to evolve stepwise therapeutic algorithms. In MOGAD, the treatment of choice remains less clear. Although some effects of rituximab as well as azathioprine can be assumed, it seems to be less effective than in NMOSD; for other therapeutic alternatives, only small case series19 20 or data in significantly younger populations are available. Further treatment studies are urgently needed, ideally in a prospective, randomised setting.
Data availability statement
Data are available upon reasonable request. The data that support the findings of this study are available from the corresponding author upon reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by Ärztekammer München Vote 424/16S. Participants gave informed consent to participate in the study before taking part.
Acknowledgments
The authors thank all patients for participating in the study and all contributors of the Neuromyelitis Optica Study Group (NEMOS) for their support.
References
Footnotes
X @NeuroVisionLab
Collaborators The Neuromyelitis Optica Study Group (NEMOS): Philipp Albrecht, Kliniken Maria Hilf, Mönchengladbach, Germany; Klemens Angstwurm, University Hospital, Regensburg, Germany; Susanna Asseyer, Charité Universitätsmedizin, Berlin, Germany; Antonios Bayas, University Hospital, Augsburg, Germany; Natalie Bednarz, University Hospital, Leipzig, Germany; Stefanie Behnke, Knappschaftsklinikum Saar, Sulzbach, Germany; Achim Berthele, Technical University, Munich, Germany; Stefan Bittner, University Medical Center of the Johannes Gutenberg University, Mainz, Germany; Franziska Bütow, Hannover Medical School, Hannover, Germany; Mathias Buttmann, Caritas-Krankenhaus, Bad Mergentheim, Germany; Eva Dawin, University Hospital, Münster, Germany; Rick Dersch, University Hospital, Freiburg, Germany; Ankelien Duchow, Charité Universitätsmedizin, Berlin, Germany; Thorleif Etgen, Kliniken Südostbayern, Traunstein, Germany; Carsten Finke, Charité Universitätsmedizin, Berlin, Germany; Katinka Fischer, University Hospital, Düsseldorf, Germany; Moritz Förster, Kliniken Maria Hilf, Mönchengladbach, Germany; Mathias Fousse, University Hospital, Homburg, Germany; Benedikt Frank, University Hospital, Essen, Germany; Frank Freitag, Nervenzentrum, Potsdam, Germany; Anna Gahlen, Ruhr University of Bochum, Germany; Achim Gass, University Hospital, Mannheim, Germany; Johannes Gehrig, University Hospital, Frankfurt, Germany; Christian Geis, Jena University Hospital, Germany; Katrin Giglhuber, Technical University, Munich, Germany; Yasemin Goereci, University Hospital, Cologne, Germany; Ralf Gold, Ruhr University of Bochum, Germany; Ana Beatriz Ayrosa Galvao Ribeiro Gomes, University Hospital, Basel, Switzerland; Jonas Graf, University Hospital, Düsseldorf, Germany; Sergiu Groppa, University Medical Center of the Johannes Gutenberg University, Mainz, Germany; Julia Gutbrod, University Hospital, Augsburg, Germany; Kerstin Guthke, Klinikum Görlitz, Germany; Axel Haarmann, University of Würzburg, Germany; Maria Hastermann, Charité Universitätsmedizin, Berlin, Germany; Kerstin Hellwig, Ruhr University of Bochum, Germany; Bernhard Hemmer, Technical University, Munich, Germany; Mariella Herfurth, University Hospital, Leipzig, Germany; Marina Herwerth, University Hospital, Zurich, Switzerland; Frank Hoffmann, Krankenhaus Martha-Maria, Halle, Germany; Olaf Hoffmann, St. Josefs-Krankenhaus, Potsdam, Germany; Ulrich Hofstadt-van Oy, Klinikum Westfalen, Dortmund, Germany; Leila Husseini, University Hospital, Göttingen, Germany; Kathleen Ingenhoven, University Hospital, Düsseldorf, Germany; Jutta Junghans, Krankenhaus Martha-Maria, Halle, Germany ; Matthias Kaste, Nordwest Hospital Sanderbusch, Sande, Germany; Karsten Kern, Knappschaftsklinikum Saar, Sulzbach, Germany; Peter Kern, Asklepios Klinik, Teupitz, Germany; Pawel Kermer, Nordwest Hospital Sanderbusch, Sande, Germany; Christoph Kleinschnitz, University Hospital, Essen, Germany; Wolfgang Köhler, University Hospital, Leipzig, Germany; Kimberly Körbel, University Hospital, Frankfurt, Germany; Markus Kowarik, University Hospital, Tübingen, Germany; Markus Krämer, Alfried-Krupp-Krankenhaus, Essen, Germany; Markus Krumbholz, University Hospital Brandenburg, Rüdersdorf, Germany; Volker Kunzmann, Nervenzentrum, Potsdam, Germany; Natalia Kurka, University Hospital, Frankfurt, Germany; Theodoros Ladopoulos, Ruhr University of Bochum, Germany; Andrea Landwehr, University Hospital, Münster, Germany; Stefan Langel, Landeskrankenhaus Rheinhessen, Germany; Ann Sophie Lauenstein, Helios Klinikum, Wiesbaden, Germany; Sarah Laurent, University Hospital, Cologne, Germany; De-Hyung Lee, University Hospital, Regensburg, Germany; Dominik Lehrieder, University of Würzburg, Germany; Frank Leypoldt, University Hospital, Kiel, Germany; Martin Liebetrau, St. Josefs-Hospital, Wiesbaden, Germany; Gero Lindenblatt, University Hospital, Düsseldorf, Germany; Ralf Linker, University Hospital, Regensburg, Germany; Lisa Lohmann, University Hospital, Münster, Germany; Peter Lüdemann, Agaplesion Ev. Bathildiskrankenhaus, Bad Pyrmont, Germany; Felix Luessi, University Medical Center of the Johannes Gutenberg University, Mainz, Germany; Michelle Maiworm, University Hospital, Frankfurt, Germany; Martin Marziniak, Isar-Amper Klinik Ost, Munich, Germany ; Christoph Mayer, Neurologischen Gemeinschaftspraxis im Bienenkorbhaus, Frankfurt, Germany; Stefanie Meister, University Hospital, Rostock, Germany; Arthur Melms, Facharztpraxis für Neurologie und Psychiatrie, Stuttgart, Germany; Mathias von Mering, Klinikum Bremen-Nord, Bremen, Germany; Imke Metz, University Hospital, Göttingen, Germany; Sven Meuth, University Hospital, Düsseldorf, Germany; Jasmin Naumann, Knappschaftsklinikum Saar, Sulzbach, Germany; Marjan Nenkov, University Hospital, Regensburg, Germany; Oliver Neuhaus, SRH Krankenhaus, Sigmaringen, Germany; Tradite Neziraj, University Hospital, Basel, Switzerland; Moritz Niederschweiberer, Charité Universitätsmedizin, Berlin, Germany; Sabine Niehaus, Klinikum Dortmund, Germany; Frederike Cosima Oertel, Charité, Universitätsmedizin, Berlin, Germany; Carolin Otto, Charité Universitätsmedizin, Berlin, Germany; Florence Pache, Charité Universitätsmedizin, Berlin, Germany; Thivya Pakeerathan, Ruhr University of Bochum, Germany; Sandra Paryjas, University Medical Center of the Johannes Gutenberg University, Mainz, Germany; Marc Pawlitzki, University Hospital, Düsseldorf, Germany; Sulyn Pepping, Charité Universitätsmedizin, Berlin, Germany; Steffen Pfeuffer, University Hospital, Giessen, Germany; Mosche Pompsch, Alfried-Krupp-Krankenhaus, Essen, Germany; Roxanne Alice Pretzsch, University Hospital, Basel, Switzerland; Anne- Katrin Proebstel, University Hospital, Basel, Switzerland; Maria Protopapa, University Medical Center of the Johannes Gutenberg University, Mainz, Germany; Hans-Ulrich Puhlmann, Schlosspark-Klinik, Berlin, Germany; Refik Pul, University Hospital, Essen, Germany; Sebastian Rauer, University Hospital, Freiburg, Germany; Torsten Rehfeldt, Dietrich Bonhoeffer Klinikum, Neubrandenburg, Germany; Nele Retzlaff, University Hospital, Rostock, Germany; Arne Riedlinger, Asklepios Klinik, Teupitz, Germany; Paulus Rommer, Medical University of Wien, Austria; Kevin Rostásy, Vestische Caritas-Kliniken, Datteln, Germany; Veith Rothhammer, University Hospital, Erlangen, Germany; Lioba Rückriem, MediClin Hedon-Klinik, Lingen (Ems), Germany; Klemens Ruprecht, Charité Universitätsmedizin, Berlin, Germany; Christoph Ruschil, University Hospital, Tübingen, Germany; Carina Saggau, University Hospital, Kiel, Germany; Muriel Schraad, University Medical Center of the Johannes Gutenberg University, Mainz, Germany; Matthias Schwab, Jena University Hospital, Jena, Germany; Patricia Schwarz, University Hospital, Tübingen, Germany; Maria Seipelt, University Hospital, Marburg, Germany; Jörn Peter Sieb, Helios Hanseklinikum, Stralsund, Germany; Gilberto Soloroza, Charité Universitätsmedizin, Berlin, Germany; Claudia Sommer, University Hospital, Würzburg, Germany; Alexander Stefanou, Katharinenhospital Stuttgart, Germany; Andrea Steinbrecher, Helios Klinikum, Erfurt, Germany; Heike Stephanik, University Hospital, Magdeburg, Germany; Verena Steuerwald, University Hospital, Augsburg, Germany; Muriel Stoppe, University Hospital, Leipzig, Germany; Klarissa Stürner, University Hospital, Kiel, Germany; Marie Süße, University Hospital, Greifswald, Germany; Athanasios Tarampanis, University Hospital, Düsseldorf, Germany; Simone Tauber, University Hospital, Aachen, Germany; Daria Tkachenko, Hannover Medical School, Hannover, Germany; Thanos Tsaktanis, University Hospital, Erlangen, Germany; Hayrrettin Tumani, University Hospital, Ulm, Germany; Ulrike Wallwitz, Krankenhaus Martha-Maria, Halle, Germany; Anna Walz, Hannover Medical School, Hannover, Germany; Klaus-Peter Wandinger, University Medical Center Schleswig-Holstein Campus, Lübeck, Germany; Martin S. Weber, University Hospital, Göttingen, Germany; Jens Weise, Helios Vogtland-Klinikum, Plauen, Germany; Jonathan Wickel, Jena University Hospital, Germany; Heinz Wiendl, University Hospital, Münster, Germany; Alexander Winkelmann, University Hospital, Rostock, Germany; Felix Wohlrab, Charité Universitätsmedizin, Berlin, Germany; Yavor Yalachkov, University Hospital, Frankfurt, Germany; Uwe Zettl, University Hospital, Rostock, Germany; Ulf Ziemann, University Hospital, Tübingen, Germany; Frauke Zipp, University Medical Center of the Johannes Gutenberg University, Mainz, Germany.
Contributors VH, TF and JPS (guarantor) contributed to the conception and design of the study; VH, CT, DE, HP, JH, AD, PS, CS, TP, KF, MR, GL, MWH, DT, FB, KG, MF, IS, MKK, SJ, ED, LR, MS, AW, MP, IK, KA, MK, MG, JW, PSR, JPS, MK, FTB, HT, LK, BW, OA, IA, JBS, FP, TK, TF, AB and JPS contributed to the acquisition, analysis of data and reviewing the manuscript. VH, AB and JPS contributed to the drafting the text or preparing the figures.
Funding This project was partially funded by Alexion AstraZeneca Rare Disease, Roche and NEMOS e.V. and supported by a grant from Innovationsausschuss of the German Federal Joint Committee (G-BA; project NUTSEN) to AB, OA, MWH, FP and CT.
Competing interests VH has received research support from NEMOS e.V. for this project. CT received honoraria for consultation and expert testimony from Alexion Pharma Germany GmbH. None of this interfered with the current report. DE received speaker honoraria and/or travel reimbursement from Alexion, Amgen/Horizon and Merck, all not related to the presented work. DE received speaker honoraria from Alexion. HP has nothing to disclose. JH reports grants from the Friedrich-Baur-Stiftung, Merck and Horizon, personal fees and non-financial support from Alexion, Horizon, Roche, Merck, Novartis, Biogen, BMS and Janssen and non-financial support from the Guthy-Jackson Charitable Foundation and the Sumaira Foundation. AD has received research support from NEMOS e.V. independent of this project. PS received travel support from UCB, received speaker‘s honoraria from Roche and Alexion and served on advisory boards by Alexion. CS has received speaker honoraria from Alexion and travel support from Novartis and UCB. TP has nothing to disclose. KF has nothing to disclose. MR received speaker honoraria and travel reimbursement from Roche, Alexion and Horizon, not related to this study. GL has nothing to disclose. MWH received institutional research support from Myelitis e. V., the German Federal Joint Committee/Innovation Fund, and NEMOS e. V. Speaker honoraria from selpers og, AMGEN/Horizon, and Alexion, and travel grants and compensation for serving on an advisory board from Alexion. DT has nothing to disclose. FB has nothing to disclose. KG has nothing to disclose. MF has nothing to disclose. IS has nothing to disclose. MKK speaker honoraria from BMS, Novartis, Merck. SJ has nothing to disclose. ED has nothing to disclose. LR has nothing to disclose. MS has received consulting and/or speaker honoraria from Alexion, Bayer, Biogen, Bristol-Myers-Squibb/Celgene, Janssen, Merck, Horizon, Roche and Sanofi Genzyme. None of this interfered with the current report. MH has nothing to disclose. AW has nothing to disclose. MP has nothing to disclose. IK has received personal compensation for consulting, serving on a scientific advisory board, speaking or other activities with Alexion, Almirall, Bayer, Biogen, GlaxoSmithKline, Hexal, Horizon, Merck, Neuraxpharm, Roche/Chugai and Sanofi. IK is the editorial board member of BMC Neurology, Frontiers in Neurology and Frontiers in Immunology. KA received travel support from Alexion, Bayer, BiogenIdec, MerckSerono, Novartis, Teva and honoria from Biogen and TEVA; he supported neuroimmunological studies for Alexion, Bayer, BiogenIdec, MerckSerono, Roche and Novartis. MK has nothing to disclose. MG has received consulting and/or speaker honoraria from Biogen, BMS, JJ, Merck, Novartis, Roche, Sanofi Genzyme and Teva. JW has nothing to disclose. PSR has received honoraria for lectures/consultancy from Alexion (Astra Zeneca), Allmiral, Amicus, Biogen, Genzyme, Horizon (AMGEN), Merck, Novartis, Roche and Teva served von advisory boards for Alexion (Astra Zeneca), Amicus, Genzyme, Horizon (AMGEN), Merck, Roche and Sandoz and received research grants from Amicus, Roche, Merck and the Austrian Science Fund (FWF). JPS has nothing to disclose. MK has received honoraria from Novartis Pharma, Chugai Pharma and Roche Pharma for presentations unrelated to the topic of this article. FTB has received, over his academic career, research support and travel grants to attend scientific meetings through his institution from the German Science Fund (DFG), German Federal Ministry of Education and Science (BMBF), Bayer-Schering, Diamed, Fresenius, Merck, Novartis, Pfizer, Roche, Sanofi and Teva; speaker fees and compensation for advisory boards from Actelion, Alexion, Bayer, Biogen, CSL Behring, Fresenius, Horizon, Merck, Novartis, Roche, Sanofi-Genzyme, Takeda and Teva. None of these are related to this work. HT received institutional research support and/or consulting/speaker honoraria from Alexion, Bayer, Biogen, Bristol-Myers Squibb, Celgene, Diamed, Fresenius, Fujirebio, GlaxoSmithKline, Horizon, Janssen-Cilag, Merck, Novartis, Roche, Sanofi Genzyme and TEVA. His research is also funded by the Ministry of Education and Research (BMBF), the Ministry of Science, Research and Arts Baden Württemberg (MWK-BW), the German Society of Multiple Sclerosis (DMSG), DMS-Stiftung, AMSEL-Stiftung, Bayern-DMSG and Chemische Fabrik Karl Bucher GmbH. LK received compensation for serving on Scientific Advisory Boards for Alexion, Biogen, Bristol-Myers Squibb, Genzyme, Horizon, Janssen, Merck Serono, Novartis, Roche and Viatris. She received speaker honoraria and travel support from Argenx, Bayer, Biogen, Bristol-Myers Squibb, Genzyme, Grifols, Merck Serono, Novartis, Roche, Santhera and Teva. She receives research support from the German Research Foundation, the IZKF Münster, IMF Münster, Biogen, Immunic AG, Novartis and Merck Serono. BW received grants from the German Ministry of Education and Research, Deutsche Forschungsgemeinschaft, Dietmar Hopp Foundation and Klaus Tschira Foundation, grants and personal fees from Merck, Novartis, Roche and personal fees from Alexion, INSTAND, Roche. OA reports grants from the German Ministry of Education and Research (BMBF) and the German Research Foundation (DFG); grants and personal fees from Biogen and Novartis; and travel support and personal fees from Alexion, Almirall, MedImmune, Merck Serono, Roche, Sanofi, Viela Bio/Horizon Therapeutics and Zambon. IA has received research support from Diamed and Chugai, speaking honoraria, travel grants and compensation for serving on a scientific advisory board from Alexion, Horizon, Roche, Merck and sanofi-aventis/Genzyme, all unrelated to this study. JB-S has received institutional research support from NEMOS e.V., Alexion and Bayer AG, personal compensation from Alexion; speaking honoraria and travel grants from Bayer Healthcare, Horizon/Amgen, Novartis and sanofi-aventis/Genzyme, in addition, compensation for serving on a scientific advisory board of Alexion, Roche and Merck, all unrelated to the presented work. FP provided research support to Neurosciences Clinical Research Center, German Ministry for Education and Research (BMBF), Deutsche Forschungsgemeinschaft (DFG), Einstein Foundation, Guthy Jackson Charitable Foundation, EU FP7 Framework Program, Biogen, Genzyme, Merck Serono, Novartis, Bayer, Roche, Parexel and Almirall, received honoraria for lectures, presentations, speakers from Guthy Jackson Foundation, Bayer, Biogen, Merck Serono, Sanofi Genzyme, Novartis, Viela Bio, Roche, UCB, Mitsubishi Tanabe and Celgene, in addition received compensation for serving on a scientific advisory board of Celgene, Roche, UCB and Merck, is an Academic Editor PLos One and Associate Editor von Neurology Neuroimmunology and Neuroinflammation, all unrelated to the presented work. TK has received speaker honoraria and/or personal fees for advisory boards from Novartis Pharma, Roche Pharma, Alexion/Astra Zeneca, Horizon Therapeutics/Amgen, Merck, Chugai Pharma and Biogen. The institution she works for has received compensation for serving as a member of a steering committee from Roche. Furthermore, she is a site principal investigator in several randomised clinical trials and her institution has received compensation for clinical trials from Novartis Pharma, Roche Pharma and Sanofi Genzyme; all outside the present work. TF reports personal fees from Aslan, Bayer, BiosenseWebster, Bristol Myers Squibb, CSL Behring, Enanta, Fresenius Kabi, Galapagos, Immunic, IQVIA, Janssen, KyowaKirin, Lilly, LivaNova, Minoryx, Novartis, Recordati, Relaxera, Roche, Servier, Viatris, VICO Therapeutics and Vifor for statistical consultancies including data monitoring committees, all outside the submitted work. AB receives funding from the Innovationsausschuss of the German Federal Joint Committee (G-BA; grant 01VSF23040) and the German Federal Ministry of Education and Research (BMBF; grant 01ZZ2102B). He has received consulting and/or speaker fees from Alexion, Argenx, Biogen, Horizon/Amgen, Merck, Novartis, Roche and Sandoz/Hexal, and his institution has received compensation for clinical trials from Alexion, Biogen, Merck, Novartis, Roche and Sanofi Genzyme; all outside the present work. J-PS has nothing to disclose.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.